Спонгиформните енцефалопатии (прионни заболявания) са тези заболявания, при които патологичните форми на прионните протеини участват в развитието. Ние знаем все повече и повече за прионните заболявания, но ключовите аспекти все още са неизвестни - понастоящем медицината не разполага със средства за лечение на пациенти от тези заболявания.
Спонгиформните енцефалопатии или прионни заболявания могат да се развият по време на живота, докато други възникват от наследствени генни мутации, налични от раждането. В рамките на тази група има няколко образувания, срещащи се при хората, примери за болестта на Кройцфелд-Якоб или фатално семейно безсъние.
Прионните болести отдавна са много загадъчни. За разлика от други патогени, като бактерии, вируси или гъби, те не съдържат нуклеинова киселина - прионите са направени само от протеини. Теорията за прионните заболявания е открита от С. Прузинер, това откритие е високо оценено в научната общност - през 1997 г. изследователят получава Нобелова награда за медицина. Въпреки че са изминали относително много години от раждането на концепцията за приона, някои учени все още вярват, че тя е непълна и изследват по-нататък естеството на тези състояния - някои от факторите, отговорни за спонгиформните енцефалопатии, вече са потвърдени.
Прионни болести: причини
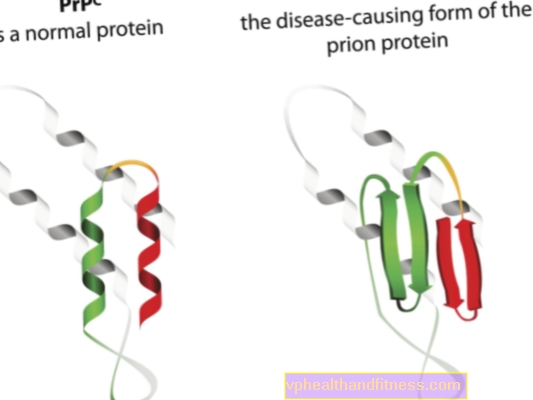
Етиологията на прионните заболявания е свързана с превръщането на нормалните прионни протеини в патогенни, патогенни форми. Прионите са протеинови молекули, които се намират в тялото на всеки човек. Тяхната функция все още не е напълно ясна, но е известно, че при нормални условия прионните протеини не увреждат тялото. Когато обаче прионите променят структурата си и се превърнат в патогенни частици, се развива една от няколко спонгиформни енцефалопатии. Прионите, срещащи се в организма, се наричат PRPC, докато ненормалните форми се наричат PRPSC. Последните са сериозен проблем не само защото могат да се натрупват в нервната тъкан под формата на отлагания и да генерират нейни увреждания, но и защото имат способността да трансформират нормалните приони в деформирана форма (просто казано, PRPSC може да "зарази" нормалните протеини с патогенния си потенциал).
Прочетете още: Болест на Хънтингтън (хорея на Хънтингтън): причини, симптоми, лечение Мускулно треперене - причини. Какво означава мускулен тремор? Болести, които убиват най-бързо: ШОК, ЕБОЛА, ПРОКЛОНА, АТАКА, АВАРИЙНОСТ [GALE ...По принцип има 3 причини за спонгиформната енцефалопатия:
- спорадични (патогенна мутация се среща в соматични клетки, възниква по време на живота на пациента),
- семейство (произтичащо от тежестта на мутациите, наследени от родители),
- Пасаж (свързан с въвеждането на патогенни приони в човешкото тяло, например чрез препарати на растежен хормон, замърсени с тези частици или трансплантация на роговица от човек, страдащ от някаква гъбеста енцефалопатия).
Спонгиформни енцефалопатии: болест на Кройцфелд-Якоб
Болестта на Кройцфелд-Якоб (CJD) е описана за първи път в началото на 20-те години. Има 4 вида заболяване:
- спорадичен CJD (най-често срещаният, отчитащ до 9/10 от всички случаи на CJD)
- родния град на CJD
- претоварени от CJD
- вариант на CJD
Клиничната картина в хода на различни варианти на болестта на Кройцфелд-Якоб е променлива. Най-честите заболявания в хода на тази група спонгиформни енцефалопатии са:
- деменция (включително прогресивно влошаване на паметта, вниманието и концентрацията)
- миоклонус (неволеви движения като внезапно изтръпване на мускулите)
- церебеларна дисфункция (проявяваща се например чрез нарушения на баланса)
- замъглено зрение
- пирамидални и екстрапирамидни симптоми
В хода на вариантите на CJD могат да се появят и психични разстройства (напр. Тревожност, депресивно настроение), болка и други неволеви движения, различни от гореспоменатите.
Прогнозата за болестта на Кройцфелд-Якоб е лоша - например при пациенти със спорадична CJD отнема средно четири до пет месеца от появата на симптомите на заболяването до смъртта.
Спонгиформни енцефалопатии: синдром на Gerstmann-Straussler-Scheinker
Синдромът на Gerstmann-Straussler-Scheinker (GSS) обикновено протича в семейства и се причинява от наследствена мутация в гена PRNP. Счита се за най-бавно прогресиращата спонгиформна енцефалопатия. Екипът на GSS включва:
- спиноцеребеларна атаксия
- дизартрия
- нарушения на деменцията
- нарушения на преглъщането
- нистагъм
- повишено мускулно напрежение
Пациентите с диагноза GSS имат различно време и при някои пациенти смъртта настъпва повече от 10 години след началото.
Спонгиформни енцефалопатии: фамилно фамилно безсъние
Фаталното фамилно безсъние е прионно заболяване, причинено от мутации в гена PRNP. Заболяването е изключително рядко и досега е диагностицирано в 28 семейства по целия свят. В хода на фаталното семейно безсъние първият симптом е невъзможността за сън. Този проблем води до тревожни разстройства и пациентът изпитва халюцинации. Ефектът от постоянната липса на нощна почивка е дисфункция на вегетативната система (включително промени в сърдечната функция, изпотяване и нарушения на храносмилателната система), има и прогресивно намаляване на телесното тегло. В по-напредналите фази на фамилното семейно безсъние се появяват хормонални нарушения и в хода на заболяването се появяват симптоми на деменция.
Прогнозата за фатално семейно безсъние, както и за други спонгиформни енцефалопатии, е лоша: пациентите обикновено умират в рамките на три години от началото.
Спонгиформни енцефалопатии: прионопатия с променлива чувствителност към протеаза
Появата на обсъжданите спонгиформни енцефалопатии е свързана главно с мутации в гена PRNP. Тези мутации обаче се отнасят до различни кодони на този ген и следователно се различават няколко различни прионни заболявания. Сравнително наскоро описана (през 2008 г.) единица е принопатията с променлива чувствителност към протеаза. Хората, страдащи от това заболяване, носят мутации в цели три кодона на гена PRNP.
Припринопатия с променлива чувствителност към протеаза пациентите изпитват:
- когнитивно увреждане
- изключителна тежест на психиатричните разстройства: те могат да бъдат еуфория и възбуда, но и значителна апатия
- дизартрия
- афазия (нарушения на езиковите функции)
Средната продължителност на заболяването при тази принопатия е по-малка от 4 години.
Спонгиформни енцефалопатии: куру
Понастоящем Куру се счита за болест, която на практика вече не съществува - тя е открита при представители на племена от Папуа Нова Гвинея, които практикуват канибалистично поведение. Доминиращият симптом на тази спонгиформна енцефалопатия е прогресираща церебеларна атаксия. То може да бъде придружено от неволни движения (главно под формата на хорея, тремор и атетоза), както и инконтиненция на урината и фекалиите. Пациентите на куру също изпитват значителни промени в настроението, те развиват примитивни рефлекси (например смучене). Доста характерен проблем в случая на тази прионна болест са принудителните пристъпи на плач или смях - поради последните явления, понякога куру се нарича „смееща се смърт“.
Спонгиформни енцефалопатии: диагноза
Прионните заболявания могат да се подозират въз основа на симптомите на пациента. Те обаче са доста неспецифични, тъй като могат да се появят и в хода на редица други заболявания, които не са свързани с прионите. Поради тази причина при диагностицирането на спонгиформни енцефалопатии също се използват следните:
- образни тестове (напр. ядрено-магнитен резонанс, който позволява да се открият промени, свързани с дегенерацията на мозъка от прионни протеини),
- лабораторни тестове (като оценка на протеиновите концентрации в цереброспиналната течност, напр. MAP-tau, S-100 или 14-3-3 протеини),
- генетични тестове (за откриване на наличие на мутации при пациента),
- имунистохимични тестове (с използване на антитела към прион протеини).
Диагнозата може да бъде потвърдена и чрез аутопсия на мозъка, при която е възможно да се открият промени, характерни за спонгиформните енцефалопатии. Това могат да бъдат гъбести лезии, различно разпределени и с различна структура (в зависимост от конкретната болест) амилоидни плаки и невронални дефекти.
Спонгиформни енцефалопатии: лечение
Понастоящем прионните болести са нелечими - въпреки многобройните проучвания, които продължават от много години, медицината все още не разполага с лекарства, които биха могли да забавят или напълно да потиснат техния напредък. При пациенти с спонгиформни енцефалопатии се използва симптоматично лечение, което има за цел да облекчи интензивността на симптомите и да подобри максимално качеството им на живот.
Въпреки това работата по лечение на спонгиформни енцефалопатии все още продължава. Учените се опитват да използват различни методи - първият пример е генната терапия. Те биха повлияли на нуклеиновите киселини и мутациите, присъстващи в тяхната структура - целта на прилагането на генната терапия би била да неутрализира грешките в генетичния код. Друг подход е в основата на имунотерапията - работи се по създаването на антитела, чиято роля би била да елиминират патогенните приони. Друг метод, който вижда потенциала за борба с спонгиформните енцефалопатии, е лечението с използване на синтезирани протеинови молекули, които, веднъж въведени в тялото на пациента, ще неутрализират патологичните протеини.
Препоръчителна статия:
Енцефалопатии - причини, видове и симптоми













.jpg)